
 Thank you for visiting the Prather Laboratory web page. Here you will have the opportunity to learn about the people in the lab that do the great things that we publish. I’ve been very fortunate to assemble a team of great people that work well together. We also have world-wide collaborations that result in accomplishing goals that are unattainable in a single laboratory.
Thank you for visiting the Prather Laboratory web page. Here you will have the opportunity to learn about the people in the lab that do the great things that we publish. I’ve been very fortunate to assemble a team of great people that work well together. We also have world-wide collaborations that result in accomplishing goals that are unattainable in a single laboratory.
Our research is focused on early embryo development in the pig. There is a 30% loss of potential conceptuses during the first month of development. We hope that by describing the cellular and molecular program of events that occur during development of the preimplantation pig embryo the root causes of these losses can be identified and steps can be taken to reduce them. To that end, three EST projects, funded by the USDA and Monsanto, were initiated with the final goal of transcript profiling of reproductive tissues in pigs and cattle. Since then, we have adopted deep sequencing technologies to identify both transcripts and methylated regions of the genome that are unique to different stages of embryogenesis. Along the way, we used that information to improve systems of in vitro embryo production and further develop technology to mature oocytes, to activate oocytes, to in vitro fertilize oocytes, to culture oocytes and embryos, and to perform embryo transfer.
Since we have expertise working with early embryo and somatic cell nuclear transfer, we have used this technology to produce genetically modified pigs both directly in his laboratory and as part of the National Swine Resource and Research Center (http://nsrrc.missouri.edu/).  The genetic modifications that have been created include single and multiple transgenes, knockouts and a knockin. We performed the first gene knockout and knockin in swine.
The genetic modifications that have been created include single and multiple transgenes, knockouts and a knockin. We performed the first gene knockout and knockin in swine.
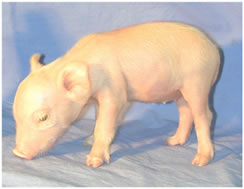
This includes creating the first miniature pigs that have the alpha 1,3 galactosyltransferase gene knocked out. This groundbreaking work has the potential to prove very useful for xenotransplantation: the transfer of pig organs into humans.
We also created pigs with a mutation in the gene that is responsible for causing cystic fibrosis (CF).  Now there is a pig model that mimics the symptoms of CF so that physicians have something to invasively experiment on and develop treatments and therapies. This is especially important since the same mutation in mice does not result in a phenotype that is similar to humans.
Now there is a pig model that mimics the symptoms of CF so that physicians have something to invasively experiment on and develop treatments and therapies. This is especially important since the same mutation in mice does not result in a phenotype that is similar to humans.
More recently we are using the CRISPR/Cas9 system to edit the genome in the in vitro produced zygote. This technology is very efficient and we have edited five genes (e.g. CD1D and CD163) and produced live normal offspring. Another five genes are in the queue.
The laboratory has made over 1,000 cloned pigs representing wild types and over 40 different genetic modifications. See the list on the History Page.